

目录
快速导航-
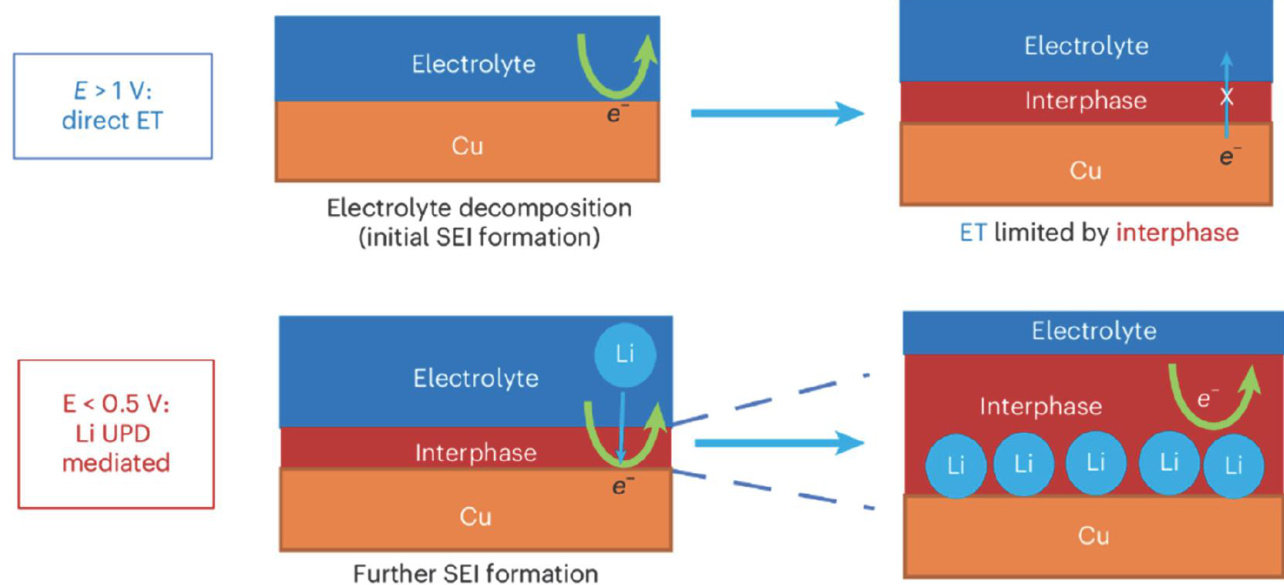
亮点 | 在锂金属电位下解析固体电解质界面膜(SEI)的化学机理
亮点 | 在锂金属电位下解析固体电解质界面膜(SEI)的化学机理
-
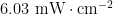
展望 | 电化学降解污水中含氮废弃物的升值策略
展望 | 电化学降解污水中含氮废弃物的升值策略
-
综述 | 锂离子电池石墨负极包覆研究进展
综述 | 锂离子电池石墨负极包覆研究进展
-
综述 | 光催化 H2O2 生产的挑战与前景
综述 | 光催化 H2O2 生产的挑战与前景
-
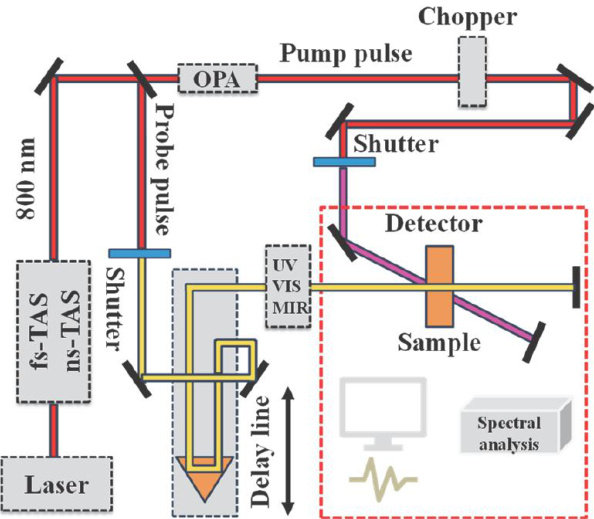
综述 | 瞬态吸收光谱在太阳能转化利用中的研究进展
综述 | 瞬态吸收光谱在太阳能转化利用中的研究进展
-
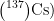
论文 | 利用碳纳米管分散的亚铁氰化锌钾电极选择性电吸附高盐放射性废水中的铯(I)
论文 | 利用碳纳米管分散的亚铁氰化锌钾电极选择性电吸附高盐放射性废水中的铯(I)
-
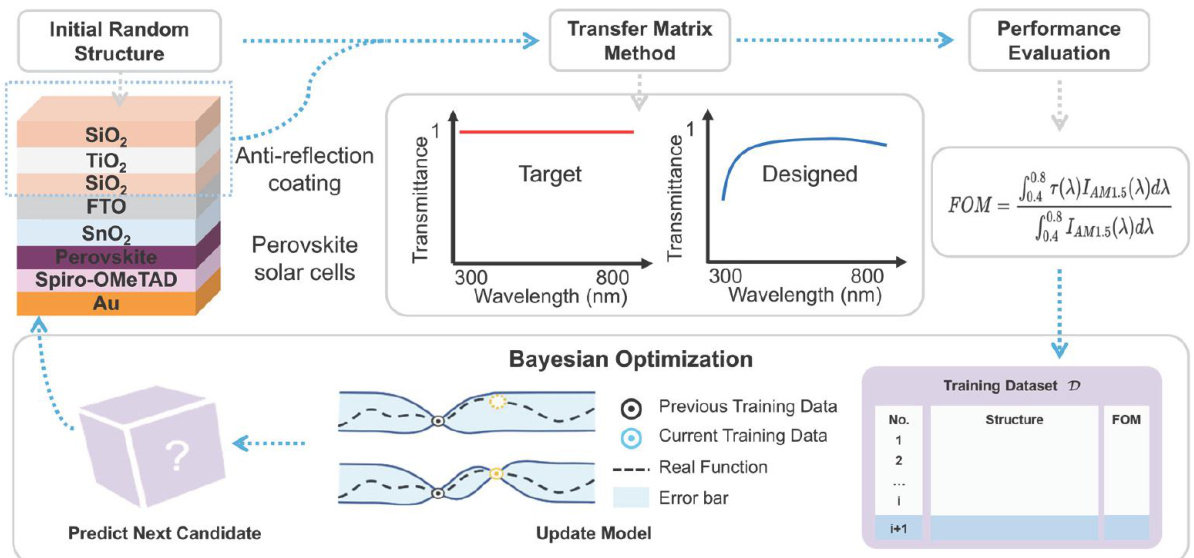
论文 | 机器学习辅助减反膜结构设计与界面修饰协同优化的高效稳定钙钛矿太阳能电池
论文 | 机器学习辅助减反膜结构设计与界面修饰协同优化的高效稳定钙钛矿太阳能电池
-
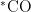
论文 | 通过边缘活性位点调控解析光热催化中析氢与二氧化碳还原的界面竞争
论文 | 通过边缘活性位点调控解析光热催化中析氢与二氧化碳还原的界面竞争
-
论文 | 单原子Pd促进的Cu催化剂用于乙醇脱氢
论文 | 单原子Pd促进的Cu催化剂用于乙醇脱氢
-
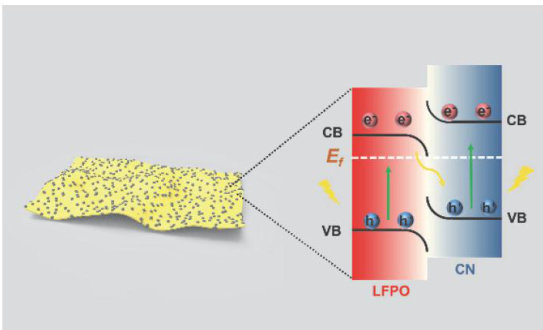
论文 | LiFePO4改善无机/有机S型光催化剂的过氧化氢光合作用
论文 | LiFePO4改善无机/有机S型光催化剂的过氧化氢光合作用
-
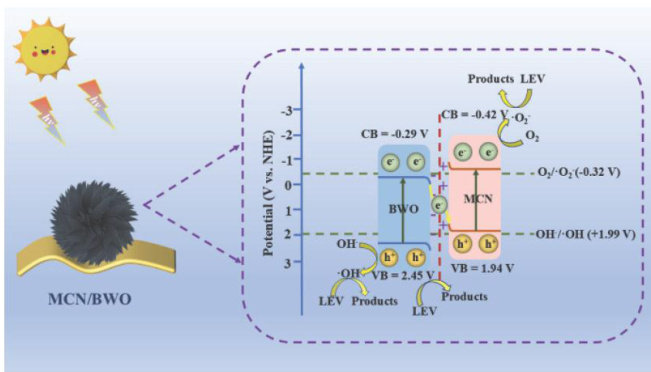
论文 | S型异质结 高效降解左氧氟沙星:性能、机理及降解路径
论文 | S型异质结 高效降解左氧氟沙星:性能、机理及降解路径
-
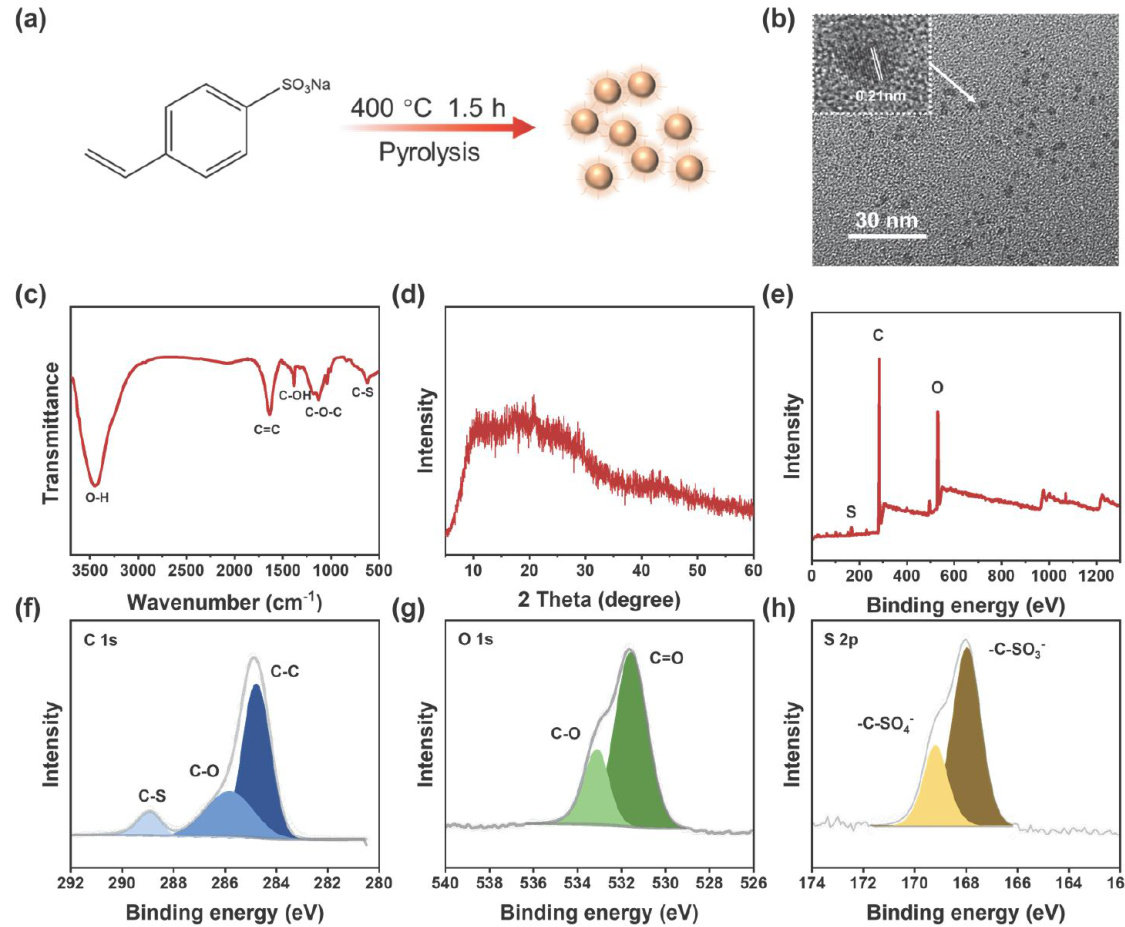
论文 | 硫掺杂的碳点作为双功能电解液添加剂实现高性能水系锌离子电池
论文 | 硫掺杂的碳点作为双功能电解液添加剂实现高性能水系锌离子电池
-
论文 | 增强 g-C3N4@BN 范德华异质结界面上的三重态电子转移增强光催化合成 H2O2
论文 | 增强 g-C3N4@BN 范德华异质结界面上的三重态电子转移增强光催化合成 H2O2
-
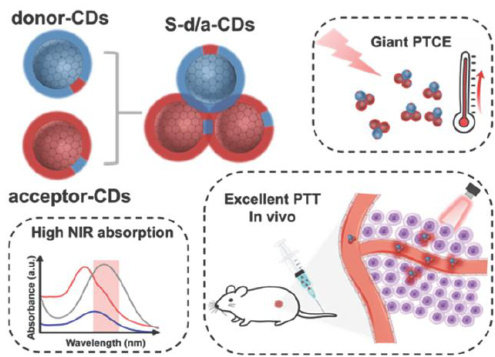
论文 | 合理组装不同表面功能化碳点以增强近红外肿瘤光热治疗效果
论文 | 合理组装不同表面功能化碳点以增强近红外肿瘤光热治疗效果
-
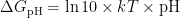
论文 | 羟基物种在过渡金属表面碱性析氢反应中的作用
论文 | 羟基物种在过渡金属表面碱性析氢反应中的作用
-
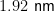
论文 | 碳量子点 IiO2 S型异质结的能带和吸附工程促进光催化 CO2 甲烷化
论文 | 碳量子点 IiO2 S型异质结的能带和吸附工程促进光催化 CO2 甲烷化
 位点协同作用降低了乙醇中C一H键断裂的活化能垒,同时增强了氢吸附和H一H键耦合能力,使得Pd/Cu-MFI催化剂获得更高的乙醇转化率和乙醛产率。
位点协同作用降低了乙醇中C一H键断裂的活化能垒,同时增强了氢吸附和H一H键耦合能力,使得Pd/Cu-MFI催化剂获得更高的乙醇转化率和乙醛产率。




















 登录
登录